
duqiang_1分析GEO数据库差异基因(GSE150910),将差异基因与泛素化酶相关基因取交集。 这个差异分析ipf chp control
但是当我使用以下代码读取时候,发现不太对读取出来的格式好像有问题 注意:观察到逗号,是不是csv的分隔符为逗号呢?3但是下载文章阅读之后 发现差异分析deseg2的时候 是需要meta信息 消除批次 年龄 实验方法这些因素影响的。莫非是因为显示的问题?在r中8-10行没有数据所以sublime或者excel查看时候,直接跳过了NA行。我知道作者为什么行名这么长了因为 有很多类型的基因都检测到了:假
·
#数据集概览
1首先geo发现 里面并没有meta信息
2 并且使用r下载之后,矩阵里面也没有meta信息。
3但是下载文章阅读之后 发现差异分析deseg2的时候 是需要meta信息 消除批次 年龄 实验方法这些因素影响的。
4但是作者说method实验方法对差异分析结果有影响
5 那么接下来,就去更原始的地方(Accession PRJNA634074; GEO: GSE150910)去找下meta信息,如果找不到,就不好办了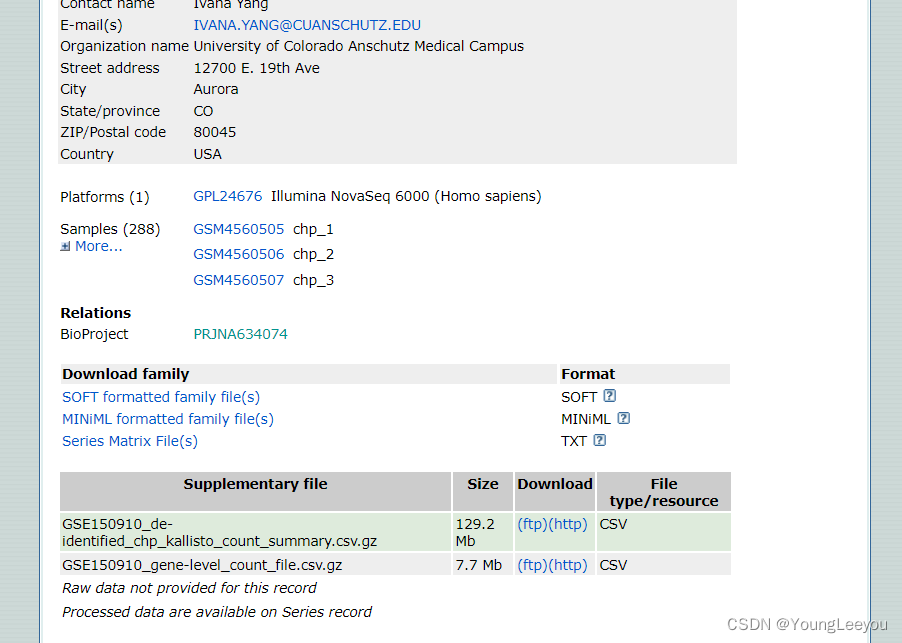
getwd()
setwd("G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/")
打开下载的矩阵csv文件,可见:
但是当我使用以下代码读取时候,发现不太对 读取出来的格式好像有问题 注意:观察到逗号,是不是csv的分隔符为逗号呢????
expr.df<-read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910__count_summary/GSE150910_de-identified_chp_kallisto_count_summary.csv",
header=TRUE,
sep="\t", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")

那么我就改变读入函数的参数 分隔符改成逗号试试呢? 结果就成功了!!!卧槽!!
expr.df<-read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910__count_summary/GSE150910_de-identified_chp_kallisto_count_summary.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")

我知道作者为什么行名这么长了 因为 有很多类型的基因都检测到了:假基因 非编码基因 编码基因
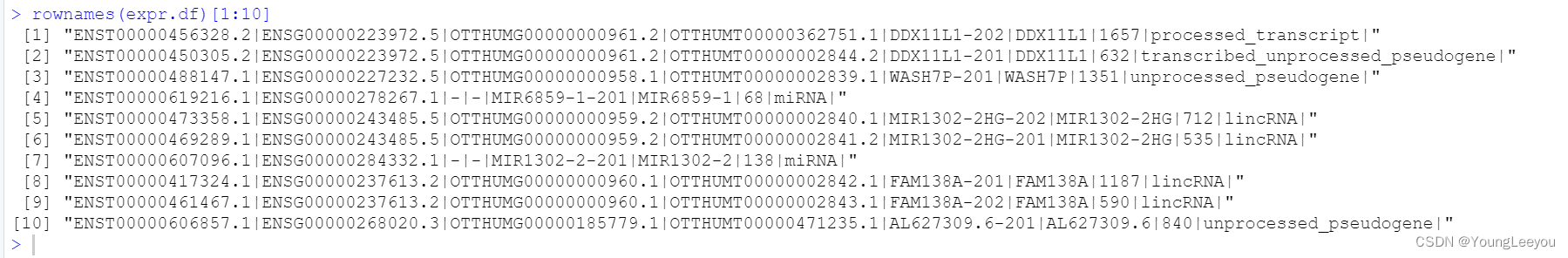
为什么会有na呢??????
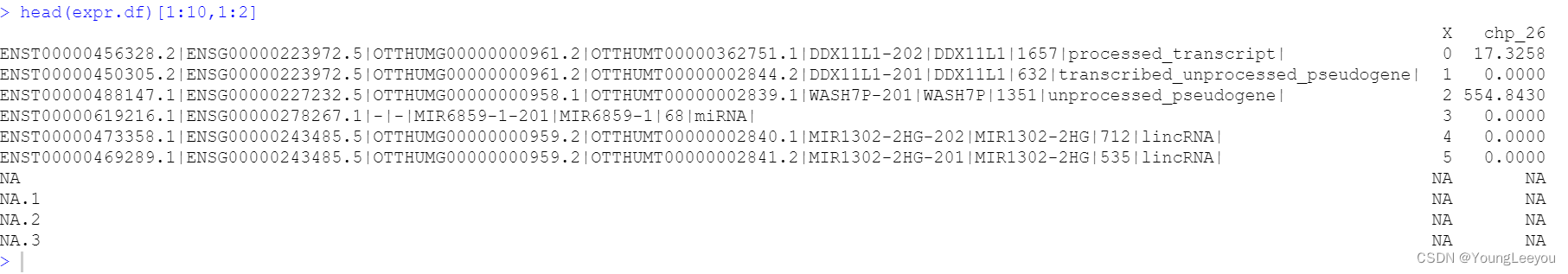
但是我使用sublime可以看到 啊 你看 6-8行都是有数据的
莫非是因为显示的问题?? 在r中8-10行没有数据 所以sublime或者excel查看时候,直接跳过了NA行
行数 在r中和在sublime软件中也是一样的
expr.df<-read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910__count_summary/GSE150910_de-identified_chp_kallisto_count_summary.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
meta_info=read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910_gene-level_count_file.csv/GSE150910_gene-level_count_file.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
if(1==1){
head(expr.df)[12:14,1:2]
head(expr.df)[2:8,1:2]
colnames(expr.df)[1:6]
rownames(expr.df)[1:10]
length(rownames(expr.df)) #200401
head(meta_info)[1:5,1:5]
boxplot(meta_info[,2:10],las=2)
match(colnames(expr.df)[-1],colnames(meta_info)[-1])#
boxplot(expr.df[,2:10],las=2)
length(rownames(meta_info))
plot(expr.df[,2:10])
??boxplot
dev.off()
ggplot2::ggplot(expr.df[,2:10])+boxplot()
#探索矩阵 发现一个基因对应多个探针
grep(pattern = 'protein',x=rownames(expr.df),value = T)[1:10]
grep(pattern ='SAMD11',x=rownames(expr.df))
expr.df[grep(pattern ='SAMD11',x=rownames(expr.df)),1:2]
#重新整合矩阵
library(dplyr)
if(1==1){grep(pattern = '|',x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",value = T)
gsub(pattern = '|',replacement = 'MM_',x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|")
sub(pattern = "|",replacement = 'MM_',x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|")
strsplit(x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",
split = "|",fixed = T) %>% as.vector()
expr.df %>% mutate(description=rownames(expr.df)) %>%
select(description,everything()) %>% head(2) %>% select(1:3)
dat=expr.df %>% mutate(description=rownames(expr.df),
name=strsplit(x=rownames(expr.df),split = "|",fixed = T)[[1]][1] ,
gene.symbol=strsplit(x=lapply(rownames(expr.df),1),split = "|",fixed = T)[[1]][6]) %>%
select(description,name,gene.symbol,everything())
for (i in rownames(expr.df)) {
norandipf.list[[i]]=RenameCells(norandipf.list[[i]],
new.names =sub(pattern = "1",
replacement =str_split(pattern = "0",
names(norandipf.list)[str_detect(names(norandipf.list),pattern = i)],
simplify = TRUE)[2],
x=colnames(norandipf.list[[i]])) )
}
library(stringr)
??str_split
str_split("ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",pattern = "|",simplify = T)
str_split("ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",pattern = "|")
for (i in rownames(expr.df)) {
print(strsplit(x=i,split = "|",fixed = T)[[1]][1])
}
myname=vector()
for (i in rownames(expr.df)) {
myname=c(myname,strsplit(x=i,split = "|",fixed = T)[[1]][1])
}
head(myname)
length(myname)
length(rownames(expr.df))
}
#自建函数,只要输入一个长的类似于"ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|"的字符串,
#就可以返回第一个 | 之前的名字
return_desired_position_value<-function(x,myposition){
strsplit(x,split = "|",fixed = T)[[1]][myposition]
}
return_desired_position_value("ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",4)
dat=expr.df
dat$description=rownames(expr.df)
dat$name=unlist( lapply(rownames(expr.df),return_desired_position_value,1))
dat$genesymbol=unlist(lapply(rownames(expr.df),return_desired_position_value,6))
rownames(dat)=seq(1,length(rownames(expr.df)),1)
head(dat)[1:4,1:9] #调整矩阵列顺序 产生新的列,并调整新列的位置
dat2=dat %>% mutate(description=rownames(expr.df),
name=unlist( lapply(rownames(expr.df),return_desired_position_value,1)),
gene.symbol=unlist(lapply(rownames(expr.df),return_desired_position_value,6)) ) %>%
select(description,name,gene.symbol,everything())
head(dat2)[1:5,1:7]
head(dat)[1:9,1:6]
head(expr.df)[1:9,1:6]
}
getwd()
。
meta_info=read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910_gene-level_count_file.csv/GSE150910_gene-level_count_file.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
head(meta_info)[1:3,1:6]
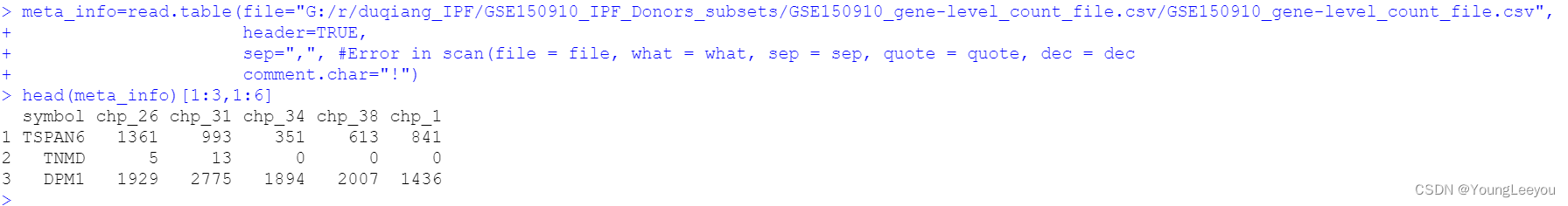
library(openxlsx)
getwd()
setwd("G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/")
expr.df<-read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910__count_summary/GSE150910_de-identified_chp_kallisto_count_summary.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
meta_info=read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910_gene-level_count_file.csv/GSE150910_gene-level_count_file.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
if(1==1){
head(expr.df)[12:14,1:2]
head(expr.df)[2:8,1:2]
colnames(expr.df)[1:6]
rownames(expr.df)[1:10]
length(rownames(expr.df)) #200401
head(meta_info)[1:5,1:5]
boxplot(meta_info[,2:10],las=2)
match(colnames(expr.df)[-1],colnames(meta_info)[-1])#
boxplot(expr.df[,2:10],las=2)
length(rownames(meta_info))
plot(expr.df[,2:10])
??boxplot
dev.off()
ggplot2::ggplot(expr.df[,2:10])+boxplot()
#探索矩阵 发现一个基因对应多个探针
grep(pattern = 'protein',x=rownames(expr.df),value = T)[1:10]
grep(pattern ='SAMD11',x=rownames(expr.df))
expr.df[grep(pattern ='SAMD11',x=rownames(expr.df)),1:2]
#重新整合矩阵
library(dplyr)
if(1==1){grep(pattern = '|',x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",value = T)
gsub(pattern = '|',replacement = 'MM_',x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|")
sub(pattern = "|",replacement = 'MM_',x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|")
strsplit(x="ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",
split = "|",fixed = T) %>% as.vector()
expr.df %>% mutate(description=rownames(expr.df)) %>%
select(description,everything()) %>% head(2) %>% select(1:3)
dat=expr.df %>% mutate(description=rownames(expr.df),
name=strsplit(x=rownames(expr.df),split = "|",fixed = T)[[1]][1] ,
gene.symbol=strsplit(x=lapply(rownames(expr.df),1),split = "|",fixed = T)[[1]][6]) %>%
select(description,name,gene.symbol,everything())
for (i in rownames(expr.df)) {
norandipf.list[[i]]=RenameCells(norandipf.list[[i]],
new.names =sub(pattern = "1",
replacement =str_split(pattern = "0",
names(norandipf.list)[str_detect(names(norandipf.list),pattern = i)],
simplify = TRUE)[2],
x=colnames(norandipf.list[[i]])) )
}
library(stringr)
??str_split
str_split("ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",pattern = "|",simplify = T)
str_split("ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",pattern = "|")
for (i in rownames(expr.df)) {
print(strsplit(x=i,split = "|",fixed = T)[[1]][1])
}
myname=vector()
for (i in rownames(expr.df)) {
myname=c(myname,strsplit(x=i,split = "|",fixed = T)[[1]][1])
}
head(myname)
length(myname)
length(rownames(expr.df))
}
#自建函数,只要输入一个长的类似于"ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|"的字符串,
#就可以返回第一个 | 之前的名字
return_desired_position_value<-function(x,myposition){
strsplit(x,split = "|",fixed = T)[[1]][myposition]
}
return_desired_position_value("ENST00000641515.1|OR4F5-202|OR4F5|2618|protein_coding|",4)
dat=expr.df
dat$description=rownames(expr.df)
dat$name=unlist( lapply(rownames(expr.df),return_desired_position_value,1))
dat$genesymbol=unlist(lapply(rownames(expr.df),return_desired_position_value,6))
rownames(dat)=seq(1,length(rownames(expr.df)),1)
head(dat)[1:4,1:9] #调整矩阵列顺序 产生新的列,并调整新列的位置
dat2=dat %>% mutate(description=rownames(expr.df),
name=unlist( lapply(rownames(expr.df),return_desired_position_value,1)),
gene.symbol=unlist(lapply(rownames(expr.df),return_desired_position_value,6)) ) %>%
select(description,name,gene.symbol,everything())
head(dat2)[1:5,1:7]
head(dat)[1:9,1:6]
head(expr.df)[1:9,1:6]
}
getwd()
#save(dat2,file = "G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910__count_summary/expr_df.rds" )
load(file = "G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910__count_summary/expr_df.rds")
meta_info=read.table(file="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/GSE150910_gene-level_count_file.csv/GSE150910_gene-level_count_file.csv",
header=TRUE,
sep=",", #Error in scan(file = file, what = what, sep = sep, quote = quote, dec = dec
comment.char="!")
head(meta_info)[1:3,1:6]
expr.df=meta_info
rownames(expr.df)=meta_info$symbol
head(expr.df)[1:8,1:5]
expr.df=expr.df[,-1]
colnames(expr.df)
expr.df=expr.df[,grep(pattern = "control|ipf",x=colnames(expr.df))]
#调整列的位置
library(dplyr)
exprdf2=expr.df %>% select(grep(pattern = "control",x=colnames(expr.df)),everything())
head(exprdf2)[1:9,1:6]
head(exprdf2)[,1:6]
exprdf2[1:9,1:6]
colnames(exprdf2)
length(rownames(exprdf2))
if (1==!1){for (eachrow in 1:length(rownames(exprdf2)) ) {
if ( is.na(mean(exprdf2[eachrow,])) ){ exprdf2=exprdf2[-eachrow,]}
}}
exprdf2[1:9,1:6]
coldata=data.frame(group=c(rep('control',length(grep(pattern = "control",x=colnames(exprdf2))) ),
rep('ipf',length(grep(pattern = "ipf",x=colnames(exprdf2))) )
),
row.names = colnames(exprdf2))
head(coldata)
library(DESeq2)
dds <- DESeqDataSetFromMatrix(countData = exprdf2,
colData = coldata,
design= ~group)
dds <- DESeq(dds)
#设置好 到底是谁比谁
dds$group <- relevel(dds$group, ref = "control") #dds$group <- factor(dds$group, levels = c("control","ipf"))
resultsNames(dds) # lists the coefficients
res <- results(dds, name="group_ipf_vs_control")
head(res)
res$genename=rownames(res)
openxlsx::write.xlsx(res,file ="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/degs_for_ipf vs control.xlsx" )
getwd()
# or to shrink log fold changes association with condition:
res <- lfcShrink(dds, coef="condition_trt_vs_untrt", type="apeglm")
head(res)
length(rownames(res)) #18838
res=as_tibble(res)
res=res %>% filter(abs(log2FoldChange)>1 & pvalue<0.05 )
length(rownames(res)) #1962
openxlsx::write.xlsx(res,file ="G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/degs_for_ipf vs control_filtered.xlsx" )
head(res)
res_up=res %>%filter(log2FoldChange>1 & pvalue<0.05 )
head(res_up)
length(rownames(res_up)) #1388
openxlsx::write.xlsx(res_up,file = "G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/degs_for_ipf vs control_filtered_up_1388.xlsx")
res_down=res %>%filter(log2FoldChange<(-1) & pvalue<0.05 )
head(res_down)
length(rownames(res_down)) #574
openxlsx::write.xlsx(res_down,file = "G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/degs_for_ipf vs control_filtered_down_574.xlsx")
enzymes=readClipboard()
head(enzymes)
length(enzymes)
#画韦恩图
library(VennDiagram)
getwd()
setwd("G:/r/duqiang_IPF/GSE150910_IPF_Donors_subsets/")
#deg_up 上调基因 与泛素化酶的交集
venn_list <- list(
degs_up=res_up$genename,
dubs_enzymes=enzymes
)
venn.diagram(venn_list, filename = 'ipf vs control_degs_up.png', imagetype = 'png',
fill = c('red', 'blue'), alpha = 0.50, cat.col = rep('black', 2),
col = 'black', cex = 1.3, fontfamily = 'serif',
cat.cex = 1.3, cat.fontfamily = 'serif')
getwd()
#继续以上述4个分组为例,组间交集元素获得
inter <- get.venn.partitions(venn_list)
#for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = ', ')
#write.table(inter[-c(5, 6)], 'ipf vs control_degs_up.txt', row.names = FALSE, sep = '\t', quote = FALSE)
for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = ', ')
openxlsx::write.xlsx(inter[-c(5, 6)], 'ipf vs control_degs_up.xlsx', row.names = FALSE, sep = ',', quote = FALSE)
#deg_down 下调基因 与泛素化酶的交集
if(1==1){
venn_list <- list(
degs_down=res_down$genename,
dubs_enzymes=enzymes
)
venn.diagram(venn_list, filename = 'ipf vs control_degs_down.png', imagetype = 'png',
fill = c('red', 'blue'), alpha = 0.50, cat.col = rep('black', 2),
col = 'black', cex = 1.3, fontfamily = 'serif',
cat.cex = 1.3, cat.fontfamily = 'serif')
getwd()
#继续以上述4个分组为例,组间交集元素获得
inter <- get.venn.partitions(venn_list)
#for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = ', ')
#write.table(inter[-c(5, 6)], 'ipf vs control_degs_up.txt', row.names = FALSE, sep = '\t', quote = FALSE)
for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = ', ')
openxlsx::write.xlsx(inter[-c(5, 6)], 'ipf vs control_degs_down.xlsx', row.names = FALSE, sep = ',', quote = FALSE)
}
#degs 与泛素化酶的交集
if(1==1){
venn_list <- list(
degs=res$genename,
dubs_enzymes=enzymes
)
venn.diagram(venn_list, filename = 'ipf vs control_degs.png', imagetype = 'png',
fill = c('red', 'blue'), alpha = 0.50, cat.col = rep('black', 2),
col = 'black', cex = 1.3, fontfamily = 'serif',
cat.cex = 1.3, cat.fontfamily = 'serif')
getwd()
#继续以上述4个分组为例,组间交集元素获得
inter <- get.venn.partitions(venn_list)
#for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = ', ')
#write.table(inter[-c(5, 6)], 'ipf vs control_degs_up.txt', row.names = FALSE, sep = '\t', quote = FALSE)
for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = ', ')
openxlsx::write.xlsx(inter[-c(5, 6)], 'ipf vs control_degs.xlsx', row.names = FALSE, sep = ',', quote = FALSE)
}
library("org.Mm.eg.db")
k <- keys(org.Mm.eg.db, keytype = "ENTREZID")
gene.list <- select(org.Mm.eg.db, keys = k, columns = c("SYMBOL", "ENSEMBL"), keytype = "ENTREZID")## 73306 3
input<-expr.df
input$symbol<-input$mgi_symbol
head(input)
input$FeatureName.entrez = gene.list[match(input$symbol, gene.list$SYMBOL),"ENTREZID"]#在input文件的基础上添加symbol与entrezid相对应的列FeatureName.entrez
head(input)
head(expreset)
expreset=expreset[-1,]
head(expreset)
colnames(expreset)=expreset[1,]
head(expreset)
expreset=expreset[-1,]
head(expreset)
expreset$gene=expreset[,2]
expreset$ensembleid=expreset[,1]
head(expreset)
silica0_silica_6_expreset=expreset[,c("p_3_1",'p_3_2',
"s_6_1","s_6_2","s_6_3",
"gene","ensembleid")]
head(silica0_silica_6_expreset)
rownames(silica0_silica_6_expreset)=silica0_silica_6_expreset$ensembleid
head(silica0_silica_6_expreset)
silica0_silica_6_expreset=silica0_silica_6_expreset[,-c(6,7)]
head(silica0_silica_6_expreset)
head(data.matrix(silica0_silica_6_expreset))
str(silica0_silica_6_expreset)
type(silica0_silica_6_expreset)
'''
nrows=202
ncols=6
counts=matrix(runif(nrows*ncols,1,1e4),nrows)
colData=DataFrame(treatment=rep(c("chr1","chr2"),3),
row.names=LETTERS[1:6])
SummarizedExperiment(assays = list(counts),
colData = colData)
'''
silica0_silica_6_expreset=data.matrix(silica0_silica_6_expreset)
SummarizedExperiment(assays =list(counts=silica0_silica_6_expreset),
colData = DataFrame(row.names = colnames(silica0_silica_6_expreset),
treatment=c(rep("control",2),
rep("sio2_42",3)
)
)
)
myexpreset=SummarizedExperiment(assays =list(counts=silica0_silica_6_expreset),
colData = DataFrame(row.names = colnames(silica0_silica_6_expreset),
treatment=c(rep("control",2),
rep("sio2_42",3)
)
)
)
group_list=colData(myexpreset)
exprSet=assay(myexpreset)
table(group_list)
dim(exprSet)
head(exprSet)
type(exprSet)
head(exprSet)
dev.off()
# 下面代码是为了检查
if(T){
colnames(exprSet)
pheatmap::pheatmap(cor(exprSet))
group_list
tmp=data.frame(g=group_list)
tmp
rownames(tmp)=colnames(exprSet)
tmp
# 组内的样本的相似性理论上应该是要高于组间的
# 但是如果使用全部的基因的表达矩阵来计算样本之间的相关性
# 是不能看到组内样本很好的聚集在一起。
pheatmap::pheatmap(cor(exprSet),annotation_col = tmp)
dim(exprSet)
# 所以我这里初步过滤低表达量基因。
exprSet[1:5,1:4]
exprSet=exprSet[apply(exprSet,1, function(x) sum(x>1) > 2),] #得到在至少2个细胞都有表达的基因
dim(exprSet)
'''
exprSet=log(edgeR::cpm(exprSet)+1) #log转化
dim(exprSet)
apply(exprSet,1, function(x) sum(x>1) > 5)
apply(exprSet, 1,mad)
head(M)
# 再挑选top500的MAD值基因 #MAD (Median absolute deviation)绝对中位值. 中位数:统计学名词,是指将统计总体中的各个变量值按大小顺序排列起来形成一个数列,处于变量数列中间位置的变量值就称为中位数. MAD:就是先求出给定数据的中位数 (注意并非均值)然后原数列的每个值与这个中位数求出绝对差,然后新数列的中位值就是MAD
exprSet=exprSet[names(sort(apply(exprSet, 1,mad),decreasing = T)[1:500]),]
dim(exprSet)
M=cor(log2(exprSet+1))
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(M)
pheatmap::pheatmap(M,annotation_col = tmp)
# 现在就可以看到,组内样本很好的聚集在一起
# 组内的样本的相似性是要高于组间
pheatmap::pheatmap(M,annotation_col = tmp,filename = 'cor.png')
pheatmap::pheatmap(M,annotation_col = tmp)
'''
library(pheatmap)
pheatmap(scale(cor(log2(exprSet+1))))
pheatmap(scale(cor(log2(exprSet+1))),annotation_col = tmp)
}
dev.off()

一站式 AI 云服务平台
更多推荐


 1
1 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)