
2024年最全MMseqs2蛋白质序列快速高效比对工具,Linux运维架构师成长路线
最全的Linux教程,Linux从入门到精通第一份《Linux从入门到精通》466页内容简介====本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、
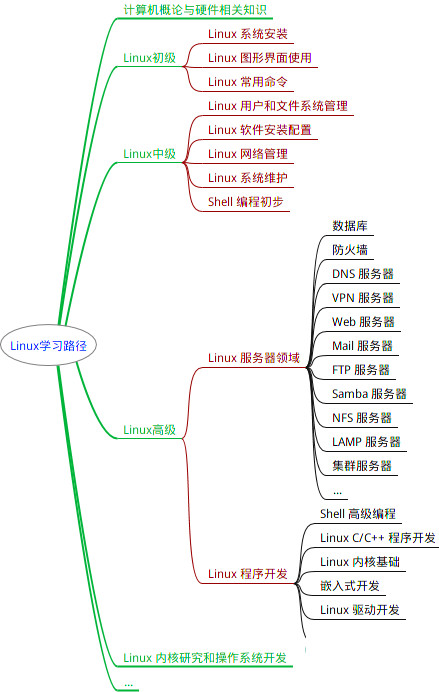
最全的Linux教程,Linux从入门到精通
======================
-
linux从入门到精通(第2版)
-
Linux系统移植
-
Linux驱动开发入门与实战
-
LINUX 系统移植 第2版
-
Linux开源网络全栈详解 从DPDK到OpenFlow

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。
需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。
一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!
# install by brew 一般是mac系统默认的,当然mac就是linux系统,所以其他linux系统也可以自己安装配置brew工具
brew install mmseqs2
# install via conda,这个大家都能用,估计做生信的都有了,直接命令安装
conda install -c conda-forge -c bioconda mmseqs2
# install docker,会容器管理的建议这个,导入导出方便,随处可移,运行完自动释放
docker pull ghcr.io/soedinglab/mmseqs2
###下面的就做参考吧,大家可能没有运维经验的会不熟悉
# static build with AVX2 (fastest)
wget https://mmseqs.com/latest/mmseqs-linux-avx2.tar.gz; tar xvfz mmseqs-linux-avx2.tar.gz; export PATH=$(pwd)/mmseqs/bin/:$PATH
# static build with SSE4.1
wget https://mmseqs.com/latest/mmseqs-linux-sse41.tar.gz; tar xvfz mmseqs-linux-sse41.tar.gz; export PATH=$(pwd)/mmseqs/bin/:$PATH
# static build with SSE2 (slowest, for very old systems)
wget https://mmseqs.com/latest/mmseqs-linux-sse2.tar.gz; tar xvfz mmseqs-linux-sse2.tar.gz;
###linux环境下就这样不用写注册表,将生成的二进制程序文件加入到系统环境中就好了。
export PATH=$(pwd)/mmseqs/bin/:$PATH
###克隆git仓库,自行编译,需要有debug经验
git clone https://github.com/soedinglab/MMseqs2.git
cd MMseqs2
mkdir build
cd build
cmake -DCMAKE_BUILD_TYPE=RELEASE -DCMAKE_INSTALL_PREFIX=. ..
make
make install
export PATH=$(pwd)/bin/:$PATH
全参数使用帮助信息:
MMseqs2 Version: 13.45111
© Martin Steinegger (martin.steinegger@snu.ac.kr)
usage: mmseqs <command> [<args>]
Easy workflows for plain text input/output
easy-search Sensitive homology search
easy-linsearch Fast, less sensitive homology search
easy-cluster Slower, sensitive clustering
easy-linclust Fast linear time cluster, less sensitive clustering
easy-taxonomy Taxonomic classification
easy-rbh Find reciprocal best hit
Main workflows for database input/output
search Sensitive homology search
linsearch Fast, less sensitive homology search
map Map nearly identical sequences
rbh Reciprocal best hit search
linclust Fast, less sensitive clustering
cluster Slower, sensitive clustering
clusterupdate Update previous clustering with new sequences
taxonomy Taxonomic classification
Input database creation
databases List and download databases
createdb Convert FASTA/Q file(s) to a sequence DB
createindex Store precomputed index on disk to reduce search overhead
createlinindex Create linsearch index
convertmsa Convert Stockholm/PFAM MSA file to a MSA DB
tsv2db Convert a TSV file to any DB
tar2db Convert content of tar archives to any DB
msa2profile Convert a MSA DB to a profile DB
Handle databases on storage and memory
compress Compress DB entries
decompress Decompress DB entries
rmdb Remove a DB
mvdb Move a DB
cpdb Copy a DB
lndb Symlink a DB
unpackdb Unpack a DB into separate files
touchdb Preload DB into memory (page cache)
Unite and intersect databases
createsubdb Create a subset of a DB from list of DB keys
concatdbs Concatenate two DBs, giving new IDs to entries from 2nd DB
splitdb Split DB into subsets
mergedbs Merge entries from multiple DBs
subtractdbs Remove all entries from first DB occurring in second DB by key
Format conversion for downstream processing
convertalis Convert alignment DB to BLAST-tab, SAM or custom format
createtsv Convert result DB to tab-separated flat file
convert2fasta Convert sequence DB to FASTA format
result2flat Create flat file by adding FASTA headers to DB entries
createseqfiledb Create a DB of unaligned FASTA entries
taxonomyreport Create a taxonomy report in Kraken or Krona format
Sequence manipulation/transformation
extractorfs Six-frame extraction of open reading frames
extractframes Extract frames from a nucleotide sequence DB
orftocontig Write ORF locations in alignment format
reverseseq Reverse (without complement) sequences
translatenucs Translate nucleotides to proteins
translateaa Translate proteins to lexicographically lowest codons
splitsequence Split sequences by length
masksequence Soft mask sequence DB using tantan
extractalignedregion Extract aligned sequence region from query
Result manipulation
swapresults Transpose prefilter/alignment DB
result2rbh Filter a merged result DB to retain only reciprocal best hits
result2msa Compute MSA DB from a result DB
result2dnamsa Compute MSA DB with out insertions in the query for DNA sequences
result2stats Compute statistics for each entry in a DB
filterresult Pairwise alignment result filter
offsetalignment Offset alignment by ORF start position
proteinaln2nucl Transform protein alignments to nucleotide alignments
result2repseq Get representative sequences from result DB
sortresult Sort a result DB in the same order as the prefilter or align module
summarizealis Summarize alignment result to one row (uniq. cov., cov., avg. seq. id.)
summarizeresult Extract annotations from alignment DB
Taxonomy assignment
createtaxdb Add taxonomic labels to sequence DB
createbintaxonomy Create binary taxonomy from NCBI input
addtaxonomy Add taxonomic labels to result DB
taxonomyreport Create a taxonomy report in Kraken or Krona format
filtertaxdb Filter taxonomy result database
filtertaxseqdb Filter taxonomy sequence database
aggregatetax Aggregate multiple taxon labels to a single label
aggregatetaxweights Aggregate multiple taxon labels to a single label
lcaalign Efficient gapped alignment for lca computation
lca Compute the lowest common ancestor
majoritylca Compute the lowest common ancestor using majority voting
Multi-hit search
multihitdb Create sequence DB for multi hit searches
multihitsearch Search with a grouped set of sequences against another grouped set
besthitperset For each set of sequences compute the best element and update p-value
combinepvalperset For each set compute the combined p-value
mergeresultsbyset Merge results from multiple ORFs back to their respective contig
Prefiltering
prefilter Double consecutive diagonal k-mer search
ungappedprefilter Optimal diagonal score search
kmermatcher Find bottom-m-hashed k-mer matches within sequence DB
kmersearch Find bottom-m-hashed k-mer matches between target and query DB
Alignment
align Optimal gapped local alignment
alignall Within-result all-vs-all gapped local alignment
transitivealign Transfer alignments via transitivity
rescorediagonal Compute sequence identity for diagonal
alignbykmer Heuristic gapped local k-mer based alignment
Clustering
clust Cluster result by Set-Cover/Connected-Component/Greedy-Incremental
clusthash Hash-based clustering of equal length sequences
mergeclusters Merge multiple cascaded clustering steps
Profile databases
result2profile Compute profile DB from a result DB
msa2result Convert a MSA DB to a profile DB
msa2profile Convert a MSA DB to a profile DB
profile2pssm Convert a profile DB to a tab-separated PSSM file
profile2consensus Extract consensus sequence DB from a profile DB
profile2repseq Extract representative sequence DB from a profile DB
convertprofiledb Convert a HH-suite HHM DB to a profile DB
Profile-profile databases
enrich Boost diversity of search result
result2pp Merge two profile DBs by shared hits
profile2cs Convert a profile DB into a column state sequence DB
convertca3m Convert a cA3M DB to a result DB
expandaln Expand an alignment result based on another
expand2profile Expand an alignment result based on another and create a profile
Utility modules to manipulate DBs
view Print DB entries given in --id-list to stdout
apply Execute given program on each DB entry
filterdb DB filtering by given conditions
swapdb Transpose DB with integer values in first column
prefixid For each entry in a DB prepend the entry key to the entry itself
suffixid For each entry in a DB append the entry key to the entry itself
renamedbkeys Create a new DB with original keys renamed
Special-purpose utilities
diffseqdbs Compute diff of two sequence DBs
summarizetabs Extract annotations from HHblits BLAST-tab-formatted results
gff2db Extract regions from a sequence database based on a GFF3 file
maskbygff Mask out sequence regions in a sequence DB by features selected from a GFF3 file
convertkb Convert UniProtKB data to a DB
summarizeheaders Summarize FASTA headers of result DB
nrtotaxmapping Create taxonomy mapping for NR database
extractdomains Extract highest scoring alignment regions for each sequence from BLAST-tab file
countkmer Count k-mers
光看帮助会有点懵了,但总体还是清晰的,下面大家可以在逐步使用中熟悉这些参数的使用方法。
这里说一下主要工作流程模块:
###帮助文件最上面是关于主要工作流程模块的介绍。
easy-search Sensitive homology search,高敏感度同源基因搜索
easy-linsearch Fast, less sensitive homology search,较低敏感度同源基因搜索
easy-cluster Slower, sensitive clustering,较慢的较高敏感度聚类
easy-linclust Fast linear time cluster, less sensitive clustering,快速线性时间聚类,低灵敏度聚类
easy-taxonomy Taxonomic classification,物种注释
easy-rbh Find reciprocal best hit,查找最佳命中
#####使用时很简单,分别查看帮助文件
mmseqs easy-search --help
mmseqs easy-linsearch --help
mmseqs easy-cluster --help
mmseqs easy-linclust --help
mmseqs easy-taxonomy --help
mmseqs easy-rbh --help
2. 下载数据库Downloading databases
#先查看有些什么数据库,可以直接使用下面的帮助信息查看
mmseqs databases
Usage: mmseqs databases <name> <o:sequenceDB> <tmpDir> [options]
Name Type Taxonomy Url
- UniRef100 Aminoacid yes https://www.uniprot.org/help/uniref
- UniRef90 Aminoacid yes https://www.uniprot.org/help/uniref
- UniRef50 Aminoacid yes https://www.uniprot.org/help/uniref
- UniProtKB Aminoacid yes https://www.uniprot.org/help/uniprotkb
- UniProtKB/TrEMBL Aminoacid yes https://www.uniprot.org/help/uniprotkb
- UniProtKB/Swiss-Prot Aminoacid yes https://uniprot.org
- NR Aminoacid yes https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA
- NT Nucleotide - https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA
- GTDB Aminoacid yes https://gtdb.ecogenomic.org
- PDB Aminoacid - https://www.rcsb.org
- PDB70 Profile - https://github.com/soedinglab/hh-suite
- Pfam-A.full Profile - https://pfam.xfam.org
- Pfam-A.seed Profile - https://pfam.xfam.org
- Pfam-B Profile - https://xfam.wordpress.com/2020/06/30/a-new-pfam-b-is-released
- CDD Profile - https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml
- eggNOG Profile - http://eggnog5.embl.de
- VOGDB Profile - https://vogdb.org
- dbCAN2 Profile - http://bcb.unl.edu/dbCAN2
- SILVA Nucleotide yes https://www.arb-silva.de
- Resfinder Nucleotide - https://cge.cbs.dtu.dk/services/ResFinder
- Kalamari Nucleotide yes https://github.com/lskatz/Kalamari
下载指定数据库
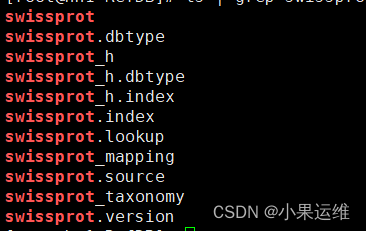
#下载swissprot数据库
mmseqs databases UniProtKB/Swiss-Prot outpath/swissprot tmp
下载完的数据库就在指定路径下,不含swissprot名, 也就是自己指定的/outpath路径,使用的时候指定数据库路径/outpath/swissprot
当然可以自己下载fasta文件手动配置数据库
3. 创建数据库
使用MMseqs创建一个数据库,该数据库将包含您要使用的蛋白质序列数据。要创建数据库,请执行以下命令:
#先将参考库fasta文件生成mmseqs对应数据库文件
mmseqs createdb <sequences.fasta> <database_name>
## 其中,`<sequences.fasta>`是您的蛋白质序列文件名,`<database_name>`是您要为数据库指定的名称。
#######################################################################################
mmseqs createdb examples/QUERY.fasta queryDB
mmseqs createdb examples/DB.fasta targetDB
4. 训练数据库
为了提高比对质量,可以训练数据库。要训练数据库,请执行以下命令:
#建立索引,加速比对
mmseqs createindex <database_name> <index_prefix>
# 其中,`<database_name>`是您之前创建的数据库名称,`<index_prefix>`是用于索引的前缀。
5. 进行比对
现在,您可以使用MMseqs比对您的蛋白质序列了。要进行比对,请执行以下命令:
mmseqs search <query.fasta> <database_name> <result_file> <tmp_dir>
#其中,`<query.fasta>`是您要比对的蛋白质序列文件名,`<database_name>`是您之前创建的数据库名称,`<result_file>`是将保存结果的文件名,`<tmp_dir>`是用于临时文件的目录。
#### 例如,这里直接用easy-search模块基于swissprot数据库进行QUERY.fasta输入文件的比对
#### 比对结果放入alnRes.m8
#### 个人建议输入文件,数据库文件还有输出文件和tmp目录统一都使用绝对路径
mmseqs easy-search examples/QUERY.fasta swissprot alnRes.m8 tmp
###结果是不是很熟悉:
k141_759496_length_1110_cov_3.0000_1 A8BQB4 0.258 337 187 0 117 369 1084 1420 2.200E-12 73
k141_759496_length_1110_cov_3.0000_1 Q2PQH8 0.258 337 187 0 117 369 1084 1420 3.903E-12 72
k141_759496_length_1110_cov_3.0000_1 P35574 0.252 337 188 0 117 369 1106 1442 6.921E-12 72
k141_759496_length_1110_cov_3.0000_1 P35573 0.244 337 191 0 117 369 1083 1419 1.205E-10 68
k141_759496_length_1110_cov_3.0000_1 Q06625 0.345 83 51 0 117 195 1067 1149 8.270E-08 59

最全的Linux教程,Linux从入门到精通
======================
1. **linux从入门到精通(第2版)**
2. **Linux系统移植**
3. **Linux驱动开发入门与实战**
4. **LINUX 系统移植 第2版**
5. **Linux开源网络全栈详解 从DPDK到OpenFlow**

第一份《Linux从入门到精通》466页
====================
内容简介
====
本书是获得了很多读者好评的Linux经典畅销书**《Linux从入门到精通》的第2版**。本书第1版出版后曾经多次印刷,并被51CTO读书频道评为“最受读者喜爱的原创IT技术图书奖”。本书第﹖版以最新的Ubuntu 12.04为版本,循序渐进地向读者介绍了Linux 的基础应用、系统管理、网络应用、娱乐和办公、程序开发、服务器配置、系统安全等。本书附带1张光盘,内容为本书配套多媒体教学视频。另外,本书还为读者提供了大量的Linux学习资料和Ubuntu安装镜像文件,供读者免费下载。

**本书适合广大Linux初中级用户、开源软件爱好者和大专院校的学生阅读,同时也非常适合准备从事Linux平台开发的各类人员。**
> 需要《Linux入门到精通》、《linux系统移植》、《Linux驱动开发入门实战》、《Linux开源网络全栈》电子书籍及教程的工程师朋友们劳烦您转发+评论
**网上学习资料一大堆,但如果学到的知识不成体系,遇到问题时只是浅尝辄止,不再深入研究,那么很难做到真正的技术提升。**
**[需要这份系统化的资料的朋友,可以点击这里获取!](https://bbs.csdn.net/forums/4f45ff00ff254613a03fab5e56a57acb)**
**一个人可以走的很快,但一群人才能走的更远!不论你是正从事IT行业的老鸟或是对IT行业感兴趣的新人,都欢迎加入我们的的圈子(技术交流、学习资源、职场吐槽、大厂内推、面试辅导),让我们一起学习成长!**

一站式 AI 云服务平台
更多推荐


 5
5 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)